

Most SDS PAGE sample buffers contain the following: SDS (sodium dodecyl sulphate, also called lauryl sulphate), -mercaptoethanol (BME), bromophenol blue, glycerol, and Tris-glycine at pH 6.8. BME is added to prevent oxidation of cysteines and to break up disulfide bonds. Bromophenyl blue is a dye that is useful for visualizing your sample in the well and tracking its progress through the gel. Glycerol is much more dense than water and is added to make the sample fall to the bottom of the sample well rather than just flow out and mix with all the buffer in the upper reservoir. The interesting components are the buffer and the SDS.
SDS-PAGE (sodium dodecyl sulphate-polyacrylamide gel electrophoresis) is commonly used in the lab for the separation of proteins based on their molecular weight. It’s one of those techniques that is commonly used but not frequently fully understood. So let’s try and fix that.
SDS-PAGE separates proteins according to their molecular weight, based on their differential rates of migration through a sieving matrix (a gel) under the influence of an applied electrical field.
The movement of any charged species through an electric field is determined by it’s net charge, it’s molecular radius and the magnitude of the applied field. But the problem with natively folded proteins is that neither their net charge nor their molecular radius is molecular weight dependent. Instead, their net charge is determined by amino acid composition i.e. the sum of the positive and negative amino acids in the protein and molecular radius by the protein’s tertiary structure.
To separate proteins in an electrical field based on their molecular weight, we need to destroy the tertiary structure by reducing the protein to a linear molecule, and somehow mask the intrinsic net charge of the protein. That’s where SDS comes in.
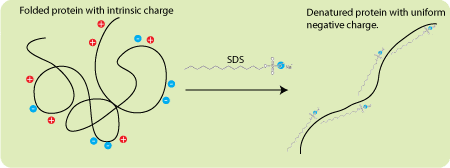
SDS is an ionic detergent that binds to the vast majority of proteins at a constant ratio of 1.4 gm SDS/gm protein. A few proteins like tubulin do not bind at this ratio and this is one reason why some proteins migrate anomalously (there are other reasons as well so you shouldn’t put too much faith in the apparent molecular weight estimated from an SDS PAGE gel). Since SDS is an anionic detergent it imparts a negative charge to all the proteins in your sample. More importantly, these charges swamp the inherent charge of the proteins and give every protein the same charge-to-mass ratio. Because the proteins have the same charge-to-mass ratio, and because the gels have sieving properties, mobility becomes a function of molecular weight. But what about running gels, stacking gels, electrode buffer, and all these different pHs?
The velocity of a charged particle moving in an electric field is directly proportional to the field strength and the charge on the molecule and is inversely proportional to the size of the molecule and the viscosity of the medium. Adding a gel with sieving properties (that is a gel where the resistance to the motion of a particle increases with particle size) increases the differences in mobility between proteins of different molecular weights. This is the basis of separation. The problem now becomes how to line up all the proteins in an orderly fashion at the starting gate. That’s where the discontinuous pH part comes in.

Laemmli gels are composed of two different gels (stacker and running gel), each cast at a different pH. In addition, the gel buffer is at a third, different pH. The running gel is buffered with Tris by adjusting it to pH 8.8 with HCl. The stacking gel is also buffered with Tris but adjusted to pH 6.8 with HCl. The sample buffer is also buffered to pH 6.8 with Tris HCl (note all the chloride ions – they will become important in a minute). The electrode buffer is also Tris, but here the pH is adjusted to a few tenths of a unit below the running gel (in this case 8.3) using only glycine – nothing else. We run our gels at constant voltage.
So here’s what happens when you turn on the power. Glycine is a weak acid and it can exist in either of two states, an uncharged zwitterion, or a charged glycinate anion (that is to say, negatively charged). At low pH it is protonated and thus uncharged. At higher pH it is negatively charged. When the power goes on the glycine ions in the running buffer want to move away from the cathode (the negative electrode) so they head toward the sample and the stacking gel. The pH there is low and so they lose a lot of their charge and slow down. Meanwhile, in the stacker and sample the highly mobile chloride ions (which are also negatively charged) move away from the cathode too. This creates a narrow zone of very low conductance (in other words very high electrical resistance) in the top of the stacking gel. Because V=IR almost all of the voltage that you put across the gel (110 Volts is typical for stacking) is concentrated in this small zone. The very high field strength makes the negatively charged proteins move forward. The trick, however, is that they can never outrun the chloride ions. If they did they would find themselves in a region of high conductance and very low field strength and would immediately slow down. The result is that all the proteins move through the stacker in a tight band just behind the moving front of chloride ions. Behind them, the pokey glycine ions straggle along as best they can (they do move, but with lower mobility than the chloride ions).
The effect of this moving zone of high voltage is that all the proteins reach the running gel at virtually the same time so that migration of the proteins is truly a function of molecular size and not some complicated function of how carefully you loaded the gel and when you started the voltage.
When the big caravan of ions hits the running gel everything changes. The pH goes way up and the glycine becomes deprotonated (and thus more negatively charged). The mobility of the glycine goes way up and the mobility of the proteins goes way down (due to the sieving properties of the gel). The result is that the glycine races past the protein and the proteins are no longer in a narrow zone of very high resistance (and very high electric field). They find themselves in a much more relaxed, uniform electric field where they can chill out a bit. Move at their own pace.
The gel matrix
In an applied electrical field, the SDS-treated proteins will now move toward the cathode at different rates depending on their molecular weight. These different mobilities will be exaggerated the high-friction environment of an gel matrix.
As the name suggests, the gel matrix used for SDS-PAGE is polyacrylamide, which is a good choice because is chemically inert and, crucially, can easily be made up at a variety concentrations to produce different pore sizes giving a variety of separating conditions that can be changed depending on your needs. You may remember that I previously wrote an article about the mechanism of acrylamide polymerisation previously .
Polyacrylamide gel polymerisation:
The unparalleled resolution and flexibility possible with polyacrylamide gel electrophoresis (PAGE) has led to its widespread use for the separation of proteins and nucleic acids. Gel porosity can be varied over a wide range to meet specific separation requirements. Electrophoresis gels and buffers can be chosen to provide separation on the basis of charge, size, or a combination of charge and size. The key to mastering this powerful technique lies in the polymerization process itself. By understanding the important parameters, and following a few simple guidelines, the novice can become proficient and the experienced user can optimize
separations even further. This bulletin takes a practical approach to the preparation of polyacrylamide gels. Its purpose is to provide the information required to achieve reproducible, controllable polymerization.
For those users interested only in the “bare essentials,” the Polymerization Protocols can be used as a laboratory guide.
Mechanism of Polymerization

a characteristic porosity which depends on the polymerization conditions and monomer concentrations.
Riboflavin (or riboflavin-5'-phosphate) may also be used as a source of free radicals, often in combination with TEMED and ammonium persulfate. In the presence of light and oxygen, riboflavin is converted to its leuco form, which is active in initiating polymerization. This is usually referred to as photochemical polymerization.
Purity of Gel-Forming Reagents
Acrylamide
Gel-forming reagents include the monomers, acrylamide and bis, as well as the initiators, usually ammonium persulfate and TEMED or, occasionally, riboflavin and TEMED. On a molar basis, acrylamide is by far the most abundant component in the monomer solution. As a result, acrylamide may be the primary
source of interfering contaminants (Dirksen and Chrambach 1972). Poor-quality acrylamide contains significant amounts of the following contaminants:
1. Acrylic acid — Acrylic acid is the deamidation product of acrylamide. Acrylic acid will copolymerize with acrylamide and bis, thereby conferring ion exchange properties on the resulting gel. This can lead to local pH changes in the gel and cause artifacts such as aberrant relative mobility, precipitation of some proteins and nucleic acids, streaking or smearing of bands, and run-to-run irreproducibility. In acrylamide, acrylic acid
should be below 0.001% (w/w). This is determined by direct titration, and supported by both conductivity and pH measurement.
2. Linear polyacrylamide — Contaminants with catalytic properties may cause what appears to be autopolymerization during the production, processing, or storage of marginally pure acrylamide. This results in the presence of linear polyacrylamide in the dry monomer. Linear polyacrylamide will ffect polymerization, since it serves as a nucleus for polymerization. The most important effect is the loss of reproducibility in gel porosity and relative mobilities of proteins and nucleic acids. Linear polyacrylamide is detected as water or alcohol insolubles and should be <0.005% (w/w).
3. Ionic contaminants — Ionic contaminants can include both inhibitors and accelerators of polymerization. Aside from acrylic acid, the most notable ionic contaminants are metals such as copper, which can inhibit gel polymerization. Metals can also poison enzymes, alter the relative mobility of metal binding proteins such as calmodulin, and inhibit digestion of electrophoretically purified nucleic acids by restriction and modification enzymes. Ionic contaminants are detected indirectly by their effects on chemical and photochemical polymerization, and by the conductivity of monomer solutions.
bis-Acrylamide
Bis is present in much smaller quantities than acrylamide in monomer solutions. However, improperly purified bis contains some of the same contaminants as acrylamide. These include products of autopolymerization and ionic contaminants, which have the same deleterious effects, and can be detected in the same ways, as the corresponding acrylamide contaminants.
Initiators
Chemical polymerization is initiated by ammonium persulfate, while photochemical polymerization is initiated by riboflavin (or riboflavin-5'-phosphate), or by a combination of riboflavin and ammonium persulfate. Initiation and polymerization are catalyzed by TEMED. Because polymerization is initiated by the generation of free radicals from persulfate or riboflavin, it is not surprising that these compounds are reactive, and
prone to oxidation or decomposition. The contaminants of the initiators tend to be the products of their own breakdown as well as other contaminating compounds. TEMED is subject to oxidation, which causes the gradual loss of catalytic activity. This process is greatly accelerated by contaminating oxidizing agents. TEMED that contains oxidation products is characterized by a yellow color. The practical consequences of the oxidative process are the requirement for greater amounts of TEMED to achieve adequate lymerization, and a gradual loss of TEMED reactivity with time. TEMED is also very hygroscopic and will gradually accumulate water, which will accelerate oxidative decomposition. TEMED with maximum activity and shelf life is obtained by redistillation immediately prior to bottling, resulting in a product that is clear, water free, and greater than 99% pure (14.4 M). Ammonium persulfate is also very hygroscopic. This property
is particularly important, since ammonium persulfate begins to break down almost immediately when dissolved in water. Therefore, the accumulation of water in ammonium persulfate results in a rapid loss of reactivity. This is why ammonium persulfate solutions should be prepared fresh daily. Persulfate is consumed in the polymerization reaction. Excess persulfate can cause oxidation of proteins and nucleic acids. This
oxidation problem can be avoided if inhibitor-free gel-forming reagents are used, and ammonium persulfate is used at the recommended levels.